ヒトの腸内マイクロバイオームで検出されたエコ進化的フィードバック
スタンフォード大学のベンジャミン・H・グッドらは、最近 Nature Communication において、ヒトの腸内マイクロバイオームの進化とそのコミュニティ組成への影響について報告しました。彼らは、マイクロバイオームが時間とともに進化できることを指摘しながらも、短期間の進化がマイクロバイオームの総合的な組成にどのような影響を与えるかはよく分かっていないと述べています。この研究では、彼らは参照ベースのアプローチを使用してマイクロバイオーム内の遺伝的変異を特定し、これらの変異がコミュニティ組成にどのような影響を与えるかを調査しています。これを探求するために、研究者たちは複数の種と宿主にわたる遺伝的比較のデータセットを分析しました。進化的イベントは種によって均等に分布していないことが分かり、進化がマイクロバイオームの生態学的組成を変化させることを示しています。また、置換と修正のイベントは、生態系構造に有意な影響を与えることから、マイクロバイオーム内に複雑なエコ進化的フィードバックが存在することを示唆しています。これは、ある種の遺伝的変化が他の種の存在量に相関する変化をもたらす可能性があることを意味しています。この研究は、腸内マイクロバイオータの短期進化を理解することが、マイクロバイオーム全体の組成を理解するための重要な意味を持つことを明らかにしました。研究所見は、マイクロバイオータの構造と機能が局所的な進化の歴史によって影響を受ける可能性があり、それはパーソナライズドメディシンやマイクロバイオーム工学に対する示唆をもたらすかもしれません。したがって、彼らは、マイクロバイオーム内の遺伝的変異によって引き起こされるコミュニティ組成の変化を調査し、この複雑な生態系内での潜在的なエコ進化的フィードバックを探求することを目指しました。
彼らはメタゲノミックパイプラインを実行しました。メタゲノミックパイプラインは、メタゲノミックデータを解析し解釈するために使用されるコンピューティングツールです。それは複雑な微生物コミュニティから意味のある情報を抽出するためのステップバイステップのプロセスに従います。このパイプラインには、データの前処理、品質管理、アッセンブリ、遺伝子予測、機能アノテーション、分類学的分類などの複数の段階が含まれます。データの前処理段階では、メタゲノミック配列の生データが刈りこまれ、フィルタリングされ、低品質のリードとアダプターが除去されます。次にアッセンブリステップでは、残りの高品質なリードが長い連続した配列であるコンティグに組みあげられます。その後、遺伝子予測アルゴリズムはコンティグ内の遺伝子を特定するために適用されます。予測された遺伝子は機能情報でアノテートされます。これには、既存のデータベースとの比較やそれらに想定される機能の割り当てが含まれています。さらに、分類学的分類の段階では、コンティグまたは予測された遺伝子を特定の分類学的グループに割り当て、微生物コミュニティの組成を決定します。メタゲノミックパイプラインはメタゲノミックデータを処理し分析するための構造的なアプローチを提供し、研究者が様々な環境での微生物コミュニティの遺伝的可能性と機能的能力を理解するのに役立つ洞察を与えます。それは彼らの生態的ニッチにおける微生物の複雑な相互作用と役割を理解するための貴重なツールであることが証明されています。
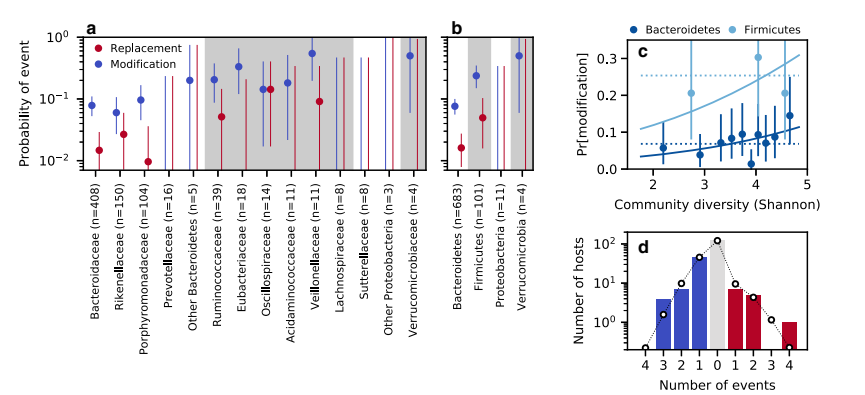
彼らは種内の置換と修正のイベントを時間を追って特定するためのデータを使用しました。彼らは主な系統が確信を持って特定できる単純な系統構造を持つ「準相フェーズ可能」な種に焦点を当てました。研究者たちは、各準相フェーズ可能集団で時点間で低頻度から高頻度へ(またはその逆)に遷移した単一ヌクレオチド変異(SNV)の数を計算しました。また、シークエンシングノイズを考慮に入れて偽陽性率を推定しました。これらのカウントに基づき、各集団は置換イベント、修正イベント、または遺伝的変化なしとしてカテゴライズされました。サンプル交換が疑われる2人の被験者は除外されました。分析では異なる種と宿主にわたり合計18回の置換イベントと98回の修正イベントが検出されました。時間間隔の重複を避けるため、データはさらにディレプリケーションされ、削減されたデータセットから16回の置換イベントと78回の修正イベントが得られました。これらの所見は後続の分析に使用されました。
研究は、在来種内進化の率が局所コミュニティの組成によってどのように影響されるかを検討しました。研究者たちは、株置換と進化的修正のイベントが税に対して均一に分布していないことを発見しました。総合的な検定により、特にファミリーまたはフィラムレベルでの種別変動の全体的な豊かさが明らかになりました。バクテロイデス門とファーミキューテス門の違いがこれらのパターンを駆動する主な役割を果たしました。ファーミキューテス門は置換と修正のイベントを高い割合で経験しています。また、研究は進化率とコミュニティ多様性の関係を調査しましたが、わずかな正の相関しか見つかりませんでした。コミュニティ多様性は健康なヒト腸内マイクロバイオームの進化率を強く制限するものではありませんでした。研究は、マイクロバイオームの遺伝的変化がグローバルなコミュニティ変数を超えた要素によって影響を受けていることを示唆し、在来種内進化の動態についての洞察を提供します。
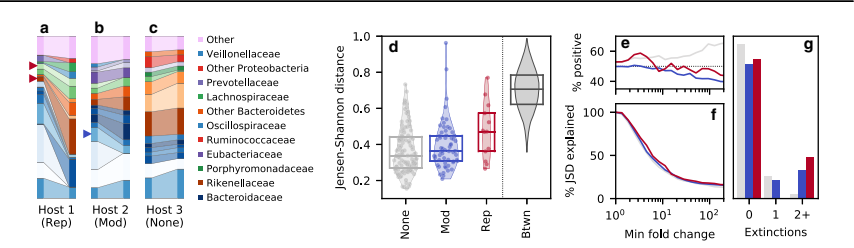
研究者たちは、バクテロイデス・ステルコリスという種で進化的修正イベントを検出しました。このイベントの間に、バクテロイデス・ステルコリスの相対的存在量がわずかに減少しました。しかし、同じ時間間隔中に2つの他の種が絶滅したことも観察されました。これらの絶滅した種のうちの1つであるバクテロイデス・マシリエンシスは、バクテロイデス・ステルコリスと同じ属に属していましたが、もう1つの絶滅した種、バルネシエラ・インテスチニホミニスは別の細菌ファミリーに属していました。これらの種の関係をより総合的に調べるため、著者たちは系統分析を行いました。彼らはJ SD(進化的修正イベントの尺度)がどれだけ注目種の同じファミリー内の種によって寄与されたかの割合を計算しました。J SDの割合を調べるとき、バクテロイデス・ステルコリスと同じファミリー内の種が有意な役割を果たしていることが、著者たちによって観察されました。この分析はこれらの種間の関係を定量的に測定するものでした。全体として、この節はバクテロイデス・ステルコリスにおいて進化的修正イベントに関する著者の所見を強調しています。相対的存在量の変化と関連種の絶滅を比較することによって、著者たちはこれらの種がどのように接続され、観察された進化的パターンへの寄与をどのようにしているかについての洞察を得ました。系統分析によって、微生物コミュニティ内の関係についてより深く理解することが可能になりました。
研究者たちは分析を行い、ヒトの腸内マイクロバイオータの短期進化とその生態構造の変化との相関を調べました。彼らは二つの可能な因果シナリオを提案しました:遺伝的変化が種間の生態学的相互作用を変える進化主導のフィードバック、および環境の撹乱が分類群のシフトをもたらし、宿主内進化の新たな機会を生み出す生態学主導のフィードバックです。これらのシナリオをよりよく理解するために、研究者たちは資源競争の単純な数学モデルを使用しました。このモデルでは、共存する種が環境によって継続的に供給される置き換え可能な資源のために競争しました。モデルは、生態学的平衡にある飽和コミュニティでは、新しい変異に対する選択圧は外部環境および在来種の豊富さに依存しないことを示しています。これは、生態学主導のフィードバックがコミュニティ全体の反応によって効果的に減衰されることを示唆しています。研究者たちはまた、変異の侵略フィットネスとそれによって引き起こされる生態学的撹乱との関係を導き出しました。この関係は、変異が系統学的または表現型的類似性にかかわらず、種の範囲にわたって正のおよび負の豊富さの変化を引き起こすことができることを示しました。これらの所見は、腸内マイクロバイオータにおける遺伝的および生態学的変化間の観察された相関を、単純な進化主導のフィードバックが説明できるという考えを支持しています。
しかしながら、研究者たちは自分たちの分析が限界を持っていることを認識しています。理論的な結果は、飽和コミュニティ内の小さな効果を持つ変異の最も単純なケースにのみ適用されました。これらの結果を飽和していないコミュニティへ拡張し、クロスフィーディングや腸内の空間構造などの他の重要な要素を組み込むために、さらなる理論的作業が必要でした。また、在来種の変異によって対応する変化をどれほど容易に生