Protéomique topographique approfondie d’une tumeur cérébrale humaine
La composition cellulaire et l’organisation spatiale des tissus jouent un rôle crucial dans la détermination de leur identité et de leur fonction. Comprendre ces caractéristiques est essentiel pour étudier les résultats des maladies. Les récentes avancées dans les technologies de séquençage spatialement résolues ont permis la caractérisation des motifs d’expression génique au sein des tissus. Cependant, pour comprendre pleinement l’hétérogénéité tissulaire, il est également nécessaire de prendre en compte les protéines codées par les gènes. Les techniques d’imagerie par spectrométrie de masse (SIM) peuvent cartographier la distribution des protéines dans les tissus, mais présentent des limitations en termes de génération de données protéomiques complètes. La microdissection avec capture laser (MCL) offre une solution prometteuse car elle permet d’extraire des régions spécifiques des tissus, allant de cellules individuelles à de plus grandes zones tissulaires.
Récemment, Simon Davis et al. d’Oxford ont utilisé la microdissection avec capture laser pour analyser la protéomique d’une tumeur cérébrale humaine à différentes résolutions spatiales, allant d’une section de tissu entière à une résolution de 40 µm. L’objectif principal était de développer un flux de travail pour la protéomique approfondie et des statistiques conscientes de l’espace afin de découvrir des caractéristiques tissulaires, plutôt que de se concentrer sur la biologie spécifique de la tumeur cérébrale. En appliquant des méthodes statistiques conscientes de l’espace, ils ont identifié des protéines et des voies avec une expression spatiale et regroupée différentielle au sein des sections tumorales, sans connaissance préalable des structures tissulaires ou de la pathologie. De plus, ils ont découvert des protéo-phénotypes définis spatialement à l’intérieur de la tumeur, révélant une biologie de la matrice extracellulaire résolue spatialement et une réponse immunitaire ciblée à la périphérie de la tumeur. De plus, ils ont cartographié les abondances protéiques par rapport à la fine structure vasculaire, fournissant des informations sur le protéome spatial dépendant des nutriments et de l’oxygène au sein d’un tissu cancéreux. Cette étude démontre le potentiel de la protéomique topographique approfondie combinée à une analyse consciente de l’espace pour identifier des zones d’intérêt liées aux mécanismes de la maladie.
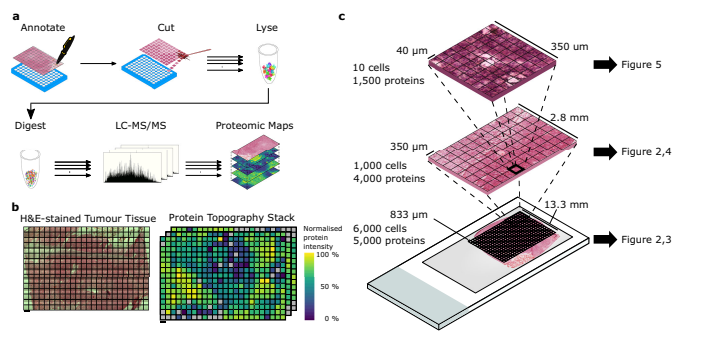
Méthodes de leur travail
Des tissus cérébraux post-mortem d’un donneur de tissus ont été obtenus avec un consentement approprié et conservés à la Banque de cerveaux d’Oxford. Le cerveau a été tranché et le tissu tumoral a été isolé des deux premières tranches coronales. Des coupes cryogéniques ont été prélevées dans différentes parties de la tumeur pour déterminer la section présentant la meilleure préservation. Le quadrant supérieur droit de la deuxième tranche coronale a été choisi pour les expériences ultérieures. Les blocs de tissus ont été préparés pour la microdissection avec capture laser en les montant sur des lames et en les colorant avec H&E.
L’analyse des tissus a été réalisée à l’aide de microscopes à microdisséquation laser équipés d’une catapultage par pression laser, plus précisément les systèmes PALM Microbeam et Leica LMD7. Le PALM Microbeam a été utilisé pour l’analyse à l’objectif 10x, tandis que le LMD7 a été utilisé pour l’analyse à l’objectif 20x. Les paramètres de coupe et de capture du Microbeam étaient Énergie : 43 et Mise au point : 55 pour la coupe, et Énergie : 20 et Mise au point : -15 pour la capture. Les échantillons collectés des deux systèmes ont été conservés à -80 °C jusqu’à leur utilisation ultérieure. Pour le système LMD7, les zones tissulaires ont été microdissectées et transférées dans des puits d’une plaque PCR de 96 puits, traitées à l’acétonitrile, puis conservées à -20 °C jusqu’à leur traitement ultérieur.
Les échantillons ont été décongelés, brièvement centrifugés et rinçés à l’aide d’un tampon RIPA contenant de la bénzonase pour collecter tout tissu restant. Les protéines ont ensuite été réduites et alkylées avant d’être mélangées avec des billes SP3 paramagnétiques. Les échantillons ont été soumis à un traitement à l’acétonitrile et secoués pour immobiliser les billes sur un aimant. Après lavage, les billes ont été remises en suspension, digérées avec de la trypsine pendant la nuit, puis soniquées. Le traitement à l’acétonitrile et l’immobilisation magnétique ont été répétés avant de transférer les peptides dans des flacons ou des plaques. Pour les données de résolution 40 µm, une digestion en une seule étape a été réalisée en utilisant un tampon de digestion contenant du n-dodécyl-β-D-maltoside et un mélange Trypsine/LysC. Les peptides obtenus ont été chargés sur des pointes C18 pour une analyse LC-MS/MS.
Les peptides des échantillons avec une résolution de 833 µm, 350 µm et 40 µm ont été analysés en utilisant différents systèmes LC-MS/MS. Pour la résolution de 833 µm, on a utilisé un Dionex Ultimate 3000 couplé à un timsTOF Pro, avec une colonne C18 de 75 μm x 150 mm et un débit de 400 nL/min. Un gradient linéaire de 17 minutes de 2% d’agent B à 30% d’agent B a été appliqué, et les spectres de masse ont été enregistrés dans la plage de 100 à 1700 m/z. Dans le cas de la résolution de 350 µm, on a utilisé un Dionex Ultimate 3000 couplé à un Orbitrap Fusion Lumos, avec une colonne C18 de 75 µm x 500 mm et un débit de 250 nL/min. Un gradient linéaire de 60 minutes de 2% d’agent B à 35% d’agent B a été appliqué, et les scans MS1 ont été acquis dans la plage de 400 à 1500 m/z. En ce qui concerne la résolution de 40 µm, on a utilisé un système Evosep One LC couplé à un spectromètre de masse timsTOF SCP, avec une colonne C18 de 75 µm x 150 mm et un débit de 1,07 secondes par cycle. Les images diaPASEF ont été séparées en 3 fenêtres de mobilité ionique couvrant la plage de masse de 400 à 1000 m/z.
Qu’ont-ils trouvé ?
Les chercheurs ont décrit leur flux de travail pour la protéomique spatialement résolue sur une tumeur téatoïde-rabdode atypique (AT/RT). Ils ont obtenu une section de la tumeur de 10 µm d’épaisseur et l’ont divisée en 384 “voxels” carrés pour l’analyse. Chaque voxel a été isolé à l’aide de la microdissection avec capture laser (MCL) et traité avec leur protocole LCMSP3. Ils ont pu identifier 5321 protéines, avec un nombre variable de protéines identifiées par échantillon, y compris des voxels vides. Les protéines identifiées ont ensuite été cartographiées à leurs positions d’origine dans la grille tissulaire. Quatre exemples de protéines ont montré différents schémas d’expression dans la section tumorale. Une analyse d’autocorrélation spatiale a été réalisée sur 4306 protéines quantifiées et 3212 protéines ont présenté une expression spatiale corrélée. Les chercheurs ont veillé à l’absence de biais d’échantillonnage et ont constaté que les différences d’abondance des protéines étaient principalement compositionnelles. Pour augmenter la résolution spatiale, ils se sont concentrés sur une région spécifique à l’intérieur de la tumeur et ont quantifié 3994 protéines dans 96 voxels plus petits. Les schémas d’expression des quatre exemples de protéines dans cette région étaient cohérents avec les données d’un champ de vision plus large. Une validation supplémentaire des données d’expression protéique spatialement résolues a été réalisée en utilisant une immunohistochimie sur trois protéines sélectionnées. Les schémas de coloration ressemblaient étroitement aux distributions d’intensité de protéines dans les cartes protéomiques.
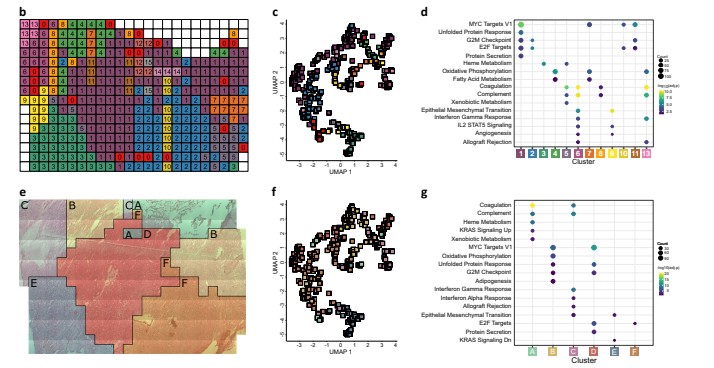
La marge entre la tumeur solide et l’interface cerveau/tumeur dans l’AT/RT a été analysée à l’aide d’une approche de regroupement qui a généré des zones spatialement définies distinctes basées sur les données de quantification des protéines. Les clusters identifiés dans cette approche étaient cohérents avec les clusters spatiaux observés dans la visualisation UMAP. Des protéines marqueurs spécifiques pour chaque cluster ont été identifiées pour étudier leurs différences fonctionnelles. Les clusters 1 et 2, qui représentent la tumeur solide, ont montré une enrichissement pour des caractéristiques prolifératives, tandis que les clusters 6 et 13, des clusters voisins dans la région supérieure gauche, ont montré une enrichissement pour des caractéristiques liées au système immunitaire. Le cluster 9, situé à la frontière de la tumeur, a montré une enrichissement en termes liés à l’angiogenèse et à la transition épithélio-mésenchymateuse, suggérant une infiltration tumorale active. L’analyse des protéines marqueurs des cellules immunitaires a révélé la localisation des marqueurs des neutrophiles et des marqueurs des macrophages M2 pro-tumoraux dans les clusters 13 et 6, respectivement. Le regroupement spatial utilisant une approche protéomique intégrée et une similarité spatiale a conduit à six clusters, englobant la tumeur solide, l’interface cerveau/tumeur, l’infiltration immunitaire et l’hémorragie. Ces clusters ont montré un enrichissement pour des caractéristiques spécifiques liées à leurs fonctions respectives. Les résultats de regroupement étaient cohérents lorsqu’ils étaient analysés à différentes résolutions spatiales. Dans l’ensemble, les clusters identifiés dans cette étude aident à définir les couches fonctionnelles à la périphérie de la tumeur dans l’AT/RT, mettant en évidence les différences d’abondance protéique et les processus fonctionnels entre la tumeur solide, l’interface et les zones environnantes.
Les chercheurs se sont concentrés sur une zone spécifique à l’intérieur du tissu tumoral contenant quatre vaisseaux sanguins représentés par un seul voxel. Ils ont utilisé une résolution spatiale plus élevée de 350 µm pour analyser les modèles d’abondance protéique et leur relation avec les gradients de nutriments et d’oxygène. La région a ensuite été divisée en une grille 9x9, donnant ainsi une résolution spatiale de 40 µm. Chaque voxel de cette taille contenait entre 5 et 10 noyaux visibles, et un total de 1550 protéines ont été quantifiées à l’aide d’une analyse indépendante des données. Les chercheurs ont mesuré la distance de chaque cellule au vaisseau sanguin le plus proche et ont constaté une corrélation positive entre les protéines sanguines et la proximité des vaisseaux sanguins. Après un regroupement spatial, le tissu a été segmenté en quatre clusters en fonction de leur proximité avec les vaisseaux sanguins. L’analyse d’enrichissement a révélé que le cluster 1, plus éloigné des vaisseaux, présentait une enrichissement du terme “Phosphorylation oxydative”, tandis que les clusters 2 et 4, plus proches des vaisseaux, présentaient une enrichissement de termes liés au sang. Les chercheurs ont observé une intensité constante de la protéine GAPDH dans tout le tissu. Les cartes protéomiques ont montré deux principaux schémas : des protéines corrélées positivement avec la distance voxel/vaisseau sanguin et des protéines corrélées négativement avec la distance voxel/vaisseau sanguin. Les chercheurs ont identifié plusieurs protéines, telles que l’alpha-2-macroglobuline et l’aspartate bêta, qui étaient corrélées aux vaisseaux sanguins et présentaient des distributions spatiales spécifiques. Ils ont également remarqué la présence de nombreuses protéines liées à la matrice extracellulaire, qui étaient spatialement corrélées et enrichies dans les voies liées à l’architecture de la matrice extracellulaire. Les chercheurs ont ensuite analysé l’abondance spatiale des protéines du collagène et des protéines de l’intégrine, qui présentaient une hétérogénéité à travers différentes sous-familles dans le tissu. Les protéines associées à la matrice extracellulaire présentaient un regroupement spatial plus élevé que les protéines non liées à la matrice extracellulaire. Dans l’ensemble, cette analyse spatiale a fourni des informations sur la distribution des protéines, les gradients de nutriments/oxygène et le rôle de la matrice extracellulaire dans le développement des tumeurs au sein du tissu tumoral.
Leurs conclusions
Cette étude a révélé des schémas d’abondance spatiale des protéines au sein de la tumeur et a identifié des protéines spécifiques qui servent de marqueurs spatiaux pour la frontière de la tumeur. De plus, l’étude a révélé des réseaux de protéines stimulés par la réponse immunitaire dans la matrice extracellulaire de la tumeur et a démontré le potentiel de la protéomique topographique approfondie pour redéfinir notre compréhension de la biologie et de la pathologie des tissus au niveau moléculaire. Cette étude aborde également les limites et les défis des techniques protéomiques à résolution spatiale actuelles et propose des orientations futures pour la recherche.