Eco-evolutionary feedbacks detected in the human gut microbiome
Benjamin H. Good et al. from Stanford University reported the evolution of the human gut microbiome and its impact on the community composition on Nature Communication recently. They stated that while the microbiome can evolve over time, the effects of short-term evolution on the overall composition of the microbiome are not well understood. In this study, they use a reference-based approach to identify genetic modifications within the microbiome and investigate how these modifications affect the community composition.To explore this, the researchers analyze a dataset of genetic comparisons across multiple species and hosts. They found that evolutionary events are not uniformly distributed across species, indicating that evolution can alter the ecological composition of the microbiome. Additionally, both replacement and modification events significantly impact the ecosystem structure, suggesting complex eco-evolutionary feedbacks within the microbiome. This means that genetic changes in one species may lead to correlated shifts in the abundances of other species.This study highlighted the importance of understanding the short-term evolution of the gut microbiota in order to comprehend the composition of the entire microbiome. The findings suggest that the structure and function of the microbiota may be influenced by their local evolutionary history, which could have implications for personalized medicine and microbiome engineering.Thus, they aimed to investigate the community composition changes resulting from genetic modifications within the microbiome and explore the potential eco-evolutionary feedbacks within this complex ecosystem.
They performed a metagenomic pipeline. The metagenomic pipeline is a computational tool used in analyzing and interpreting metagenomic data. It follows a step-by-step process to extract meaningful information from complex microbial communities. This pipeline involves several stages, including data preprocessing, quality control, assembly, gene prediction, functional annotation, and taxonomic classification.During the data preprocessing stage, raw metagenomic sequences are trimmed and filtered to remove low-quality reads and adaptors. Then, in the assembly step, the remaining high-quality reads are assembled into longer contiguous sequences called contigs.Next, gene prediction algorithms are applied to identify genes within the contigs. These predicted genes are then annotated with functional information, which involves comparing them to existing databases and assigning putative functions to them. Additionally, the taxonomic classification stage involves assigning the contigs or predicted genes to specific taxonomic groups to determine the composition of the microbial community.The metagenomic pipeline provides a structured approach to process and analyze metagenomic data, allowing researchers to gain insights into the genetic potential and functional capabilities of microbial communities in various environments. It has proven to be a valuable tool in understanding the complex interactions and roles of microorganisms in their ecological niches.
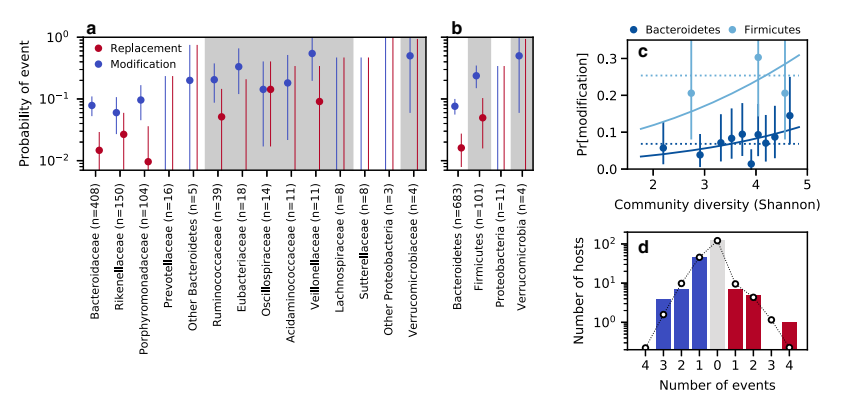
They used data to identify replacement and modification events within species over time. They focused on “quasi-phaseable” species with a simple lineage structure, allowing for the identification of dominant lineages with confidence. The researchers calculated the number of single nucleotide variants (SNVs) that transitioned from low to high frequency (or vice versa) between time points in each quasi-phaseable population. They also accounted for sequencing noise to estimate false positive rates. Based on these counts, they categorized each population as experiencing a replacement event, a modification event, or no genetic changes. Two subjects were excluded due to possible sample swapping. The analysis revealed a total of 18 replacement events and 98 modification events across different species and hosts. To avoid overlapping time intervals, the data were further de-replicated, resulting in 16 replacement events and 78 modification events from a reduced dataset. These findings were used for subsequent analyses.
The study examined how the rates of within-species evolution were affected by the composition of the local community. The researchers found that both strain replacement and evolutionary modification events were not evenly distributed among taxa. An omnibus test revealed a global enrichment of variability across species, especially at the family or phylum levels. The differences between the Bacteroidetes and Firmicutes phyla played a major role in driving these patterns, with the Firmicutes experiencing replacement and modification events at higher rates. The study also investigated the relationship between evolution rates and community diversity, but found only a weak positive correlation. Community diversity did not strongly limit the rate of evolution in healthy human gut microbiomes. The study suggests that genetic changes in the microbiome are influenced by factors beyond global community variables and provides insights into the dynamics of within-species evolution in microbial communities.
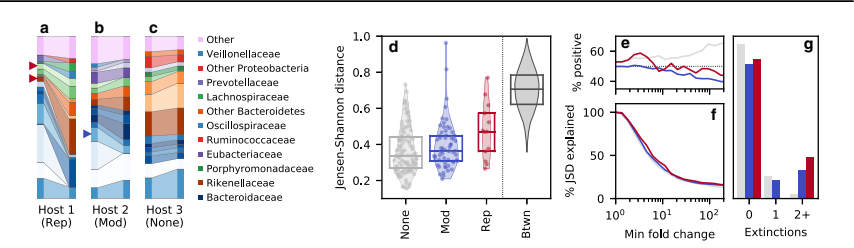
They detected an evolutionary modification event in a species called Bacteroides stercoris. During this event, the relative abundance of Bacteroides stercoris decreased slightly. However, they also observed that two other species became extinct during the same time interval. One of these extinct species, Bacteroides massiliensis, belonged to the same genus as Bacteroides stercoris, while the other extinct species, Barnesiella intestinihominis, belonged to a different bacterial family.To examine the relationships between these species more comprehensively, the authors conducted a systematic analysis. They calculated the fraction of J SD (a measure of the evolutionary modification event) that was contributed by species within the same family as one of the focal species.They found that when examining the fraction of J SD, the authors observed that species within the same family as Bacteroides stercoris played a significant role. This analysis provided a quantitative measure of the relationships between these species.Overall, this section highlights the authors’ findings regarding an evolutionary modification event in Bacteroides stercoris. By comparing the changes in relative abundance and the extinction of related species, the authors gained insight into how these species are connected and how they contribute to the observed evolutionary patterns. The systematic analysis allowed for a more in-depth understanding of the relationships within the microbial community.
The researchers conducted an analysis to investigate the correlation between the short-term evolution of the human gut microbiota and changes in its ecological structure. They proposed two possible causal scenarios: evolution-driven feedbacks, where genetic changes alter ecological interactions between species, and ecology-driven feedbacks, where environmental perturbations lead to taxonomic shifts and create new opportunities for within-host evolution.To gain a better understanding of these scenarios, the researchers used a simple mathematical model of resource competition. In this model, coexisting species competed for substitutable resources that were continuously supplied by the environment. The model showed that in a saturated community at ecological equilibrium, the selection pressures on new mutations were independent of the external environment and the abundances of the resident species. This suggested that ecology-driven feedbacks were effectively damped by the collective response of the community.The researchers also derived a relationship between the invasion fitness of a mutation and the ecological perturbations it caused after invading. This relationship showed that mutations could lead to a mixture of positive and negative abundance changes across a range of species, regardless of their phylogenetic or phenotypic similarity. These findings supported the idea that simple evolution-driven feedbacks could explain the observed correlations between genetic and ecological changes in the gut microbiota.
However, the researchers acknowledged that their analysis had limitations. The theoretical results only applied to the simplest case of small-effect mutations in saturated communities. Further theoretical work was needed to extend these results to non-saturated communities and incorporate other important factors like cross-feeding and spatial structure within the gut. Additionally, more information was needed on the genetic architectures of the resource uptake phenotypes to determine how easily corresponding changes could be produced by mutations in the resident strains.
Overall, the study provided insights into the potential mechanisms behind the observed correlations between genetic and ecological changes in the human gut microbiota. Further experiments and theoretical work were needed to fully understand these mechanisms and map out the accessible ecological perturbations caused by mutations.